寻找我儿子的生命杀手
译者按:本文作者为美国犹他(Utah)大学计算机系副教授Matthew Might,他的儿子患了一种及其罕见的基因病。他和妻子坚持不懈的为儿子寻找疾病的原因,终于完成了世界上首例的该病的确诊。他写这篇博文,希望对经历类似病症的人有帮助。让我感动的还有,即使面对这样的困难,他依然用如此诙谐的语言写下本文。祝福他们。
我终于找到了我儿子生病的原因,这花了我三年的时间,但是我们做到了。
喔这只是个示意图:

先澄清一点:我的儿子还很好的活着。尽管如此,我的妻子Cristina和我需要对他生病负责。
我的儿子Betrand患有一种罕见的基因病,事实上他是世界上首例确诊的病人。在确诊的过程中,杜克(Duke)大学对我,我妻子和我儿子做了全外显子组(whole-exome)测序,发现我的儿子遗传了同一个基因的两个变异——这个基因用于NGLY1(N-聚糖酶1)的编码。他因此不能制造这种酶。他也是目前(作者初次写这篇文章时——译者注)为止唯一已知的缺少这种酶的人。
下面我要记述的是我们不可思议的诊断历程,这是个关于只有科学能提供的希望的故事。
更新:这篇科学文献详细的记述了这个前所未有的诊断。
更新:从最初写了这篇文章后,我们又知道了全球范围内的另外15例NGLY1酶缺乏症患者(由外显子或者基因测序确诊)。如果你是一个家长,医生或者科学家,并且了解可能的病患,请理解这种酶缺乏症的表型/症状在病患之间呈现很大的不同,并且和性别相关。你的病患可能和这篇文章中的描述不同。如果你怀疑你身边有这样的人,请迅速和我联系。所有病人的信息都是保密的。
更新:我和另外一位家长合作写了一篇科学文章,发表在Genetics in Medicine (基因医学期刊)上,详细的记述了八位NGLY1酶缺乏症患者的症状。
本文还有以下翻译版本:
正常出生
除了重度黄疸,Betrand出生正常。
出生两个月内,他发育正常。
三个月时,他的发育已经放缓,但还是“正常变化范围内”。
六个月时,他几乎没有运动控制能力。
我们觉得他似乎有点“乱动”。
一定是什么事情出错了。
脑损伤
Betrand八个月大的时候第一次看发育儿科医生 - 在我们刚刚搬到犹他之后。
他做检查的那天,我在参加我的第一个教师退休会,我发现我妻子给我发了大量的语音邮件和短信。
我的心跳加速。
儿科医生认为Bertrand有脑损伤,所以她预约了后面一周的核磁共振检查。
没有脑损伤
MRI显示了一个看起来健康,正常的大脑。
所以,他的案例被提升到了小儿神经科医师那里。
神经科医生证实,他有运动障碍,但其表征“令人费解”:看起来既不象“不随意运动(Chorea)”,也不象“共济失调(Ataxia)”。
神经科医生需要验血检查。
这仅仅是几十次验血的开始。
(我们现在给Bertrand的最爱的抽血护士寄节日贺卡。)
第一次判死刑
这次抽血报告的结果只有一个异常:相对于他的年龄来说的异常高的甲胎蛋白(AFP)。
只有少数已知的紊乱会导致甲胎蛋白升高。
已知的运动障碍和AFP升高的交集只有:共济失调毛细血管扩张症(AT)。
AT是一种退化性的,致命的,无法治愈的,无法治疗的疾病。
我和妻子都伤心欲绝。
父母有血缘关系吗
因为AT是一种常染色体隐性遗传疾病,我和妻子常常被问道:
“你们两个确定你们没有血缘关系?”
我是俄亥俄州农民和北欧血统。我的妻子家人在波多黎各生活了好几代了。
我们翻了几个世纪的家谱,答案还是“没有”。
我们没有血缘关系。
写给计算机程序员看的基因学入门
要理解为什么医生不断的询问我们是否有血缘关系,以及Bertrand的最终诊断到底有多大概率,你需要了解基因和基因突变是怎么一回事。
DNA
你的基因组包含编译和运行你的身体所需要的信息。
你的基因组通过DNA来表达 - 这种分子编码的语言只包含四个字母:A,C,T和G。
(A,C,T和G代表腺嘌呤,胞嘧啶,胸腺嘧啶和鸟嘌呤。)
A,C,T和G对生命体而言,相当于计算机中的0和1。
对生命来说,重要的是这些序列如何将信息编码或计算。
在计算中,像00000100序列可能意味着“添加到位”,这样的话下面这个序列0000100000010010可能意味着“将在寄存器1中的数加到寄存器2中的数”。
密码子和标准遗传密码
在计算中,大多数计算机上运行x86指令集。
值得注意的是,在生命体中,也有一个占主导地位的指令集 - 标准的遗传密码,如DNA的密码子表中描述的那样。
遗传密码是一个指令集,通过将单个氨基酸链接起来制造蛋白质。
遗传密码是由称为密码子的指令组成的。
DNA中的每个密码子由三个字母组成,告诉某个氨基酸或者是接入某个蛋白质,或者是停止该蛋白质的构造。
例如,该密码子TTG指“接入一个亮氨酸”。
DNA的字母表中包含四个字母,因此理论上有43=64个可能的密码子。然而遗传指令仅有25个,因为一些密码子对同一个氨基酸进行编码,而且表示停止的指令也有不止一个。
基因
在人类基因组中,基因是一个功能单位——就像程序中的一段代码。每一个基因是由外显子(涉及表达蛋白质的密码子序列)和内含子(被忽略的序列,就像代码注释)组成的。
外显子描述了如何将氨基酸序列折叠成一个蛋白。
当一个基因被编译成蛋白质后——通常是某种酶——酶在细胞内像个函数一样起作用:酶接受输入分子,并给出输出分子。
除了男人的性染色体,人类的每个基因都有两种版本——一个来自母亲,另一个来自父亲。
有两个功能类似的基因提供了冗余。
变异和进化
基因的冗余是一个功能强大的防止基因突变的机制。
突变意味着生物体的遗传密码的改变。
一些突变改变DNA的字母,例如,T到A,或A到G。
这可以改变被接入的氨基酸,例如从TTT(苯丙氨酸)到TTA(亮氨酸),也可能不改变,例如从TTA(亮氨酸)到TTG(亮氨酸)。
停止的突变
虽然不常见,但也有可能发生这样的突变:密码子变成“停止”指令,它过早终止蛋白质的生产,例如从TGG(色氨酸)到TGA(停止)。
这种突变称为无义突变,因为由此产生的蛋白质是很少能够执行它原本要执行的任务。
帧突变
有些突变插入或删除DNA字母。如果这些字母的个数是三的倍数,那么插入或删除后的密码子可以被正确的读出,这就是所谓的同帧突变。
同帧突变可能会稍微改变蛋白质的功能。
(他们也可以完全破坏这些蛋白质功能。)
如果这个数不能被三整除,那么插入或删除后的密码子就会出现乱码。这是移码突变。
在大多数情况下,一个基因上靠后一些的突变的影响会小一些。(但是,在我儿子的案例中,即使是基因的最末端移码突变也可能破坏所产生的蛋白质。)
然后会发生什么?
当一个突变发生时,有四种可能的结果:
什么也没有发生。 一些突变不影响所得到的蛋白质的功能(或结构)。但是,即使该突变破坏了该蛋白质,可能也不会出现这样的迹象。如果从父母来的另一条冗余基因能够产生足够的这种蛋白质,那么就没有症状。大多数情况的基因突变都是这种结果。冗余太好了!
功能不足。 如果另一条冗余基因不能产生足够的蛋白质,可能会有不同程度的症状出现,从几乎难以察觉的症状,到严重甚至致命的症状都有可能出现。
造成伤害。 如果该突变基因产生的新蛋白质会以有害的方式起作用,那么症状就会出现。当单个异常基因导致出现问题时,这被称为常染色体显性遗传疾病。
进化。 如果该突变基因产生更好的,更有效的蛋白质,那么新的个体是更健壮的。其生存和繁殖的概率会增大。
常染色体隐性遗传性疾病
常染色体隐性遗传性疾病,例如上文中提到的AT,常常是无害的(当然这条基因本身不能正常工作)。然而如果和这条隐性基因配对的另一条基因也突变了, 就会导致问题出现。
如果携带同一个突变基因的两个后代结合,每个人都携带这条突变基因的一个拷贝,那么他们的某个孩子有四分之一的机会继承两条突变的基因。
这就是为什么许多遗传性疾病都与特定的群体或地域相关联的——其中一个共同的祖先的突变增加了其可能性。
当有人继承了两个同样的突变基因,父母常常是(非常遥远)的亲戚。
不过,我妻子和我显然没有这个问题。
即使如此,Bertrand还是有一种常染色体隐性遗传疾病。
父亲是生父吗
Bertrand长的比较象他妈妈,这一点显然没有太多帮助。
在我们确认我们没有血缘关系之后,后来的几年里,很多医生会把我妻子拉到一边问她:你确定他是Bertrand的生父吗?
感谢你们的好意,但是您怀疑错了。
进入空集
AT诊断的震惊过后,我们开始了研究。
几天之内,我们确信,Bertrand确实没有这种疾病。
即使他有异常高的甲胎蛋白和运动障碍,他的表型和医学文献中有所不同。
我们给他做了AT的基因测试,正如我们所料,结果是阴性。
在10个月大的时候,Bertrand的症状落在了空集之上。
随着每一个新的发现,空集不断变得更空。
尽可能做最好的尝试
各种小的发现在后来几个月不断出现。
每一个发现都激起了新一轮的(关于病因的)假设和结果为阴性的测试。
我们发现异常升高的谷丙转氨酶(ALT)和谷草转氨酶(AST),这常意味着肝功能异常。
然而,全面的胃肠检查又否定了这个猜测。
Bertrand的智力发育停止了大约8个月大,直到今天仍然这样,尽管他现在已经4岁了。
Bertrand奇异的病症引起了很多专科医生的注意,并尝试去诊断,然而没有一位医生成功。
再一次被宣判死刑
在我儿子大约15个月大的时候,医生们有了更大的发现。
我们在他的尿液当中发现了低聚糖,一种单糖链。
这一发现意味着某种类型的基因异常:细胞先天性代谢缺陷。
这种类型疾病中的具体亚型包括oligosaccharidoses(暂无中文名),溶酶体贮积症,先天性糖基化疾病和线粒体疾病。
(我们现在知道,Bertrand新创建一个新的类别:先天性去糖基化疾病)
Bertrand的预期寿命被削减至大约两到三年。
我们那时不知道他得的是哪一个,但这个名单上都是些外星文一般的疾病:
Alpha-Fucosidosis
Alpha-Mannosidosis
Alpha-N-Acetylgalactosaminidase Deficiency
Aspartylglycosaminuria
Beta-Mannosidosis
Galactosialidosis
Gaucher Disease
GM1 gangliosidosis
GM2 gangliosidosis
GSD II (Glycogen Storage Disease Type II)
I-Cell Disease
Mucolipidosis II
Mucolipidosis III
Pompe Disease
Pseudo-Hurler Polydystrophy
Sandhoff Disease
Schindler Disease
Sialidosis
选择
除线粒体疾病,这些疾病的产生往往是因为不能产生一个细胞代谢过程中所需要的酶。
从理论上讲,将这种缺失的酶导入到他的体细胞内可以阻止疾病的进展。
在少数病例中,酶可以合成,但传输酶到所有细胞内是一个复杂的药学问题。
(因为很多的分子都无法越过血-脑屏障。)
但是,在大多数情况下,人类还不知道如何合成这些酶。
于是,Bertrand唯一的机会是骨髓移植。
创造基因嵌合体
在进行骨髓移植的过程中,受体的用于制造干细胞的骨髓会被杀光(不可避免的,病人自身到这个时候基本上已经不能正常工作了),捐献的骨髓会替代这些骨髓,继续制造干细胞。
干细胞可以长成任何形式的细胞,因此,他们在身体的生长和修复上起很重要的作用。
随着捐献的骨髓制造的干细胞在体内的繁殖和扩散,受体变成了几个基因的嵌合体:一个有两个基因来源的生命体。
一旦捐献的干细胞开始制造这种缺少的酶,受体的身体就会开始正常的工作了,至少会变好。
在杜克大学
在发现低聚糖的几周之内,我们开始了血液测试以缩小我们探索这种疾病(以及缺少的酶)的范围。
与此同时,我们来到杜克大学来看Joanne Kurtzberg医生,她是用骨髓干细胞移植进行先天性代谢异常疾病的治疗专家。
在给Bertrand进行冒险的干细胞移植(大致30%的死亡率),Kurtzberg医生想知道问题究竟出在什么地方。
所以,我们会见了遗传学家Vandana Shashi博士和Kelly Schoch。
我们与他们以及他们的团队合作至今。
癫痫和白质流失
在杜克大学,神经学团队做了一个脑电图和磁共振。
他们发现Bertrand的脑中有大量异常的,类似癫痫的脑电活动。
(直到今天,Bertrand的脑电图还在吸引其他人来查看。)
磁共振现实他的大脑有延迟的髓鞘化。
要么是他的大脑中的白质在流失,或者是没有跟上他的生长。从功能上说,白质是神经网络的基础设施。
这个发现和脑白质退化病吻合,后者常由细胞先天代谢异常引起。
没有选择了
我们离开杜克之前,Kurtzberg直白而充满同情的告诉我们,无论Bertrand的异常是哪种,这种异常已经发展的如此之深,骨髓移植已经没有不会有作用了。
我们要崩溃了。
专注在治疗上
从杜克大学回来之后,我们把精力从诊断转到了治疗上。
发现Bertrand的很多异常的动作都是抽搐让我们很不安。
实际上,Bertrand有三种不同类型的抽搐:肌阵挛性抽搐,失神“两眼发直”抽搐和无力型“耷拉”抽搐。
几个月之内,他开始经历长时间的无力型抽搐,伴随疼痛的全身肌肉收缩。
Bertrand开始服用Keppra(左乙拉西坦),一种广泛使用的用于治疗抽搐的处方药。
生酮饮食
我们发现Keppra只是能部分有效地控制他的抽搐,我们尝试了生酮饮食。
生酮饮食是一种严格的高脂肪的饮食,迫使大脑的能量供应从葡萄糖切换到酮体。
关于生酮饮食,每吃1克碳水化合物和/或蛋白质,Bertrand需要另外摄入4克脂肪。
这种饮食已被广泛研究并用于配方,但是对于它用于很多疑难抽搐的治疗的工作机理,大部分还是未知的。
在用上这种饮食之后,Bertrand的无力型抽搐发作几乎完全停止,其他类型的发作也大大减少。
但是,当Bertrand开始出现强直性抽搐时,我们开始寻找其他的选择。
没有眼泪
某些症状真的是太明显了,明显到我们一直都没有发现。
在Bertrand大约两岁的时候,我们意识到他从来没有流过眼泪。
他会哭。
但是从来没有泪水。
简单的搜索“无泪症”之后,我们发现了Allgrove综合症这个词条。
到此为止,我们已经测试了所有已知的与先天细胞代谢有关的疾病,所以我们很迫切的要跟上最新的研究前沿。
到NIH去
我妻子联系了NIH(美国国家健康研究所,National Institute of Health)的Stratakis医生,他是Allgrove综合症的专家。
Bertrand罕见的症状引起了他的注意,他让我妻子和儿子飞往NIH去会诊。
会诊专家组推测Bertrand并非患有Allgrove综合症,但是需要一个基因诊断来断定。
基因诊断的结果现实为阴性。Bertrand没有Allgrove综合症。
会诊专家组的最后结论是Bertrand可能有男性Rett综合症,或者可能是Schinzel-Giedion综合症。
尽管症状上和Rett综合症很相近,但是进一步的检测现实Bertrand没有这两种中的任何一种异常。
激素疗法
随着Bertrand的强直性抽搐越来越剧烈和频繁的发生,我们迫切的想停止这些抽搐。
在某些无法控制的抽搐案例中,大剂量的促肾上腺皮质激素可以控制它们。
很幸运的,这个疗法对Bertrand有用。
然而,一天两次的激素注射有副作用。
他的体重翻了倍,他的头发变得稀少,开始出现秃顶,他开始长面部毛发,他长期处于愤怒状态。
想想火箭般速度的青春期发育出现在两岁孩子的身上。
尽管如此,抽搐停止了。
濒死体验
激素疗法和生酮饮食嘎然而止。
因为激素疗法停止了Bertrand免疫系统的正常运作。
几个月后,他得了严重的呼吸道感染。
他小小的两岁身躯充积着过多的体液,他几乎无法运动。
他身上的每个部分都在肿胀,他看上去像个气球娃娃。
我们想他可能挺不过去了。
为了救他,激素疗法和生酮饮食都停了,取而代之的是一系列的管子,电线和抗生素。
他看起来像星战电影中的机械人。
欢笑
停止激素疗法和生酮饮食的第二天早上,我们听到了以前从未听到的声音:笑声。
他依然浑身鼓胀,但是他在笑。每当医院的电视上出现笑语,他就加入。
这是我们见过的Bertrand的人性最直接的表达。
我妻子潸然泪下。
暴风眼
Bertrand恢复后,他的抽搐停止了几乎两个月。
甚至他的脑电图看上去也变得正常。
没了抽搐,Bertrand开始学习和成长。
对我们家来说,这是我们最开心的两个月。
抽搐又回来了
之后,抽搐还是回来了,不过只有肌阵挛型抽搐。其他类型的抽搐没有再出现。
在用药中加入拉莫三嗪缓解了抽搐,但让他看上去蔫蔫的。
我们继续的探求正确的诊断。
肝纤维化
由于Bertrand的肝功能一直异常,我们同意给他做一个肝部组织检查。
这个检查排除了医生最近的两个忧虑:Lafora病和Unverricht Lundborg病.
不幸的是,检查发现Bertrand肝部出现纤维化。
他的胃肠专科医生预测他最终会发展出肝硬化并出现肝衰竭。他推荐熊去氧胆酸,我们同意试一试。
心脏问题:长QT间歇
在Bertrand的住院时间,他的心电图显示长QT综合征。
长QT是一种罕见的心脏疾病,可导致致命的心律不齐。
长QT通常具有遗传基础,但它也可以是药物诱发的。
我们很快开始调查先天性离子通道紊乱是否是病因。
在这段时间内,Bertrand的大脑,心脏和肝脏的疾患似乎争着要将他杀死。
危险的假设
几乎穷尽了每一个可能的途径,我妻子和我做了假设。
由于我们并没有血缘关系(因而常染色体隐性遗传疾病的出现几率很小),加上我们任何一家都没有遗传疾病的历史,我们假设,Bertrand的病症可能是新生突变的结果。
我们开始假设Bertrand,而不是我们,有一个独特的基因突变。
因此,我们认为再生另一个孩子应该没有风险。
然而我们错了。
但是,假设Bertrand真的有一个新的突变,我们酝酿了一个来找到它的计划。
和基因专家的晚餐
我约犹他大学的基因学专家Lynn Jorde博士共进晚餐。我问他有没有可能对我,我妻子和我儿子三个人都进行基因测序。
基因测序产生关于一个生命体的基因编码,对人来说,它包含大约三十一亿个字母。
根据我们的推测,如果我们有这些测序结果,我们应该能找出Bertrand和我们俩不一样的基因。
但是说起来容易做起来难。即使做了,也不容易找出来。
测序的出错概率
基因测序的过程是会出错的。这意味着,可能会出现假阳性的突变结果。
这个出错的概率大约是一万分之一,所以可能会有三十一万个假阳性的突变。
重复的测序会降低假阳性的比例,但是会使得代价升高。
在餐桌上,我估测了这个数字:要得到一个可靠的基因测序,大约需要花费五十万美元。
Jorde博士很抱歉的点头表示同意。
然后怎么办
但是,假设我们发现了突变,然后呢?
我们不得不逐一调查,看看每一个突变是如何影响蛋白质的结构和功能的,这无疑是一项艰巨的任务。
但是,从理论上讲,它还是可行的。
机会:外显子组测序
贯穿Bertrand的病程,杜克大学的Shashi博士和Kelly Schoch一直和我们保持着联系,并与我们合作来检验我们的假设。
他们有强烈的预感,Bertrand有一个未被发现的遗传性疾病。
他们设计了一个巧妙的方法来检验这一假设。
他们建议使用一种新技术——对我们家三人进行全外显子组测序,而非基因测序。
只有约2%的DNA - 称为外显子组 - 主动的参与蛋白质的编码。
据估计,在这2%的基因上出现的突变是绝大多数遗传性疾病的根源。
全外显子组测序可以提供较低成本的DNA片段测序。
如果突变是在Bertand的外显子组,我们将能够找到它。
Bertrand和其他11个未确诊的儿童,参加了在杜克大学进行的试点研究。
避开了两发子弹
通过使用熊去氧胆酸治疗肝纤维化,并且进行密切的观察,我们看到了稳步的改善。
一年之后,他所有的肝脏指标都显示为正常。
Bertrand不会死于肝衰竭。
经过重复的测试,他的心脏长QT是药物导致的,具体来说是用于治疗呼吸道感染的红霉素。
角膜侵蚀
在排除了这两个疾患之后,Bertrand出现了严重的眼部感染。
其中一只眼对抗生素没有反应,需要手术排出角膜中的脓。
没有泪水导致他的角膜受到侵蚀。
他的瞳孔下面出现伤疤,模糊了他的视力。
在眼科医生的帮助下,我们开始每隔两个小时给他的眼睛滴眼药水来润滑。
这之后他的眼睛的状况有所改善,但是即使半天忘记滴眼药水都会导致持续一周甚至更长的眼部感染和结疤。
Bertrand这时身边没有护理人员的时间不能超过几个小时。
怀孕
在外显子组测序后大约一个月,我妻子怀上了我们的第二个孩子。
我们意识到,我们有机会发现,我们未出生的孩子是否有(或可能有)和Bertrand相同的疾病。
我们决定,不管我们了解到了什么,我们不会使用这些信息来指导这次怀孕的过程。
(该实验的伦理和道德协议决定,怀孕的家庭是因为正是这个棘手的伦理问题不能参加实验。)
干细胞
在外显子组实验进行的同时,我们继续治疗Bertrand的抽搐。
其中的一个假设是,Bertrand的突变不是一个完全的突变 - 在他发育成几个细胞的时候,其中某个细胞出现了突变。
这种情况的的医学名称为体细胞嵌合体,因为这种情况下,个体基因是两个非常相似的基因源的混合。
这种可能的假设下,我们决定赌一把。
我们在Bertrand出生时,存了Bertrand的脐带血干细胞。
杜克大学的Kurtzberg医生正在参与儿童干细胞的的实验,测试干细胞输注(区别于移植)对有大脑缺陷的儿童的影响。
输注自身的干细胞是无害的,我们准备试一试。
我们的想法是,如果突变没有影响到他的脐带血,这些干细胞可能开始帮助修复一些脑损伤,并制造他缺少的酶。
即使它并不制造这些酶,我们有理由希望,它可能会暂时停止或逆转他大脑白质的损失。
去Cleveland(克利夫兰)诊所
鉴于Bertrand的抽搐有多灶性,医生告诉我们手术不是选择。
但是,为了要绝对肯定,我们前往Cleveland诊所,它是手术治疗小儿癫痫领域的领导者。
尽管克利夫兰诊所证实,Bertrand不适合手术,他们增加他的药物,显著减少他的抽搐发作。
但MRI检查有惊人的发现:Bertrand的大脑白质流失(暂时)趋于稳定。
就其本身而言,这还不能证实干细胞输注已经开始显效,但它是令人鼓舞的,它表明有必要继续进行干细胞治疗的研究。
第一个基因突变的预测
在Bertrand三岁半时,再过一个星期他的妹妹Victoria就要出生了,我们接到了杜克大学Shashi博士的来电。
由于伦理和道德协议,他们不能透露他们的发现,但他们有强烈的预感,Victoria不会受Bertrand的疾病的影响。
他们想要我岳父岳母的血液样本。
我们把事情拼在一起:杜克大学研究小组的发现可能和X染色体上的变异有关。
当X染色体上含有一个突变时,女性常常不会受到影响:她们有一个多余的X染色体来补偿。
当男人有一个突变的X染色体时,这种冗余的缺乏可引起严重的症状。
这当中的信息是,如果我儿子有一个与X染色体有关的病症,我们的女儿Victoria可能是一个基因携带者,但不会比我妻子受到的影响更大。
我们都非常高兴。
但是,杜克的小组出错了。
错误的突变
我们的女儿出生以后,我岳父岳母的血液测试结果也出来了。
我岳父在他的X染色体上有相同的基因突变。
尽管这种与X染色体有关的突变在超过一千个基因组的控制数据库中是独一无二的,它影响的某种蛋白也可能能够解释Bertrand的某些症状,但事实上,我岳父的正常表现排除了它作为主导因素的角色。
缺乏维生素
有时候,解决方案似乎是太明显了,反而被人忽视。
在Bertrand的例子中,维生素就是这么一回事。
在读尽干眼症的各种原因后,我妻子偶然发现某些维生素缺乏可能导致这种症状。
尽管Bertrand每天服用复合维生素,他的某些储存在肝脏中的维生素严重不足。
在每天服用大剂量的这些维生素后,他的这些维生素水平进入“正常”范围。
此后,他已经能哭出眼泪 - 没有太多 - 但也足够。
不幸的是,由于角膜瘢痕的存在,他不再眨眼,所以他仍然需要经常眼部软膏和润滑药水 - 但比以前少多了。
确诊:NGLY1酶缺乏
在Bertrand四岁半时,我们接到了来自杜克大学的电话。
外显子组研究结束了,他们有了答案。
我和我妻子每个人都携带一个突变的NGLY1基因。
我有一个位于外显子8上的“停止”的无义突变;我妻子在最后一个外显子上有个移码突变。
我们俩产生的NGLY1酶大约是正常人的一半。Bertrand继承了我们两个的突变基因,他完全没有能力制造这种酶。
测试结果在这里,Bertrand是上面标记的“Patient 2”,带状物的宽度显示酶的制造量:
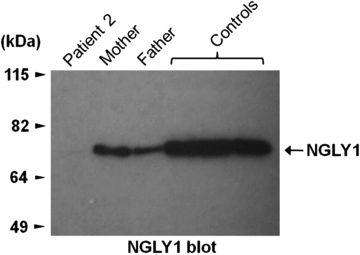
他是第一例并且是已知的唯一一例病患:
NGLY1酶在对折叠错误的蛋白质进行去糖基化的过程中具有重要作用,允许它们被回收并分解为它的组份氨基酸。
Bertrand的细胞中似乎积累了太多错折叠的糖蛋白。
几率
这个诊断值得我们思考,这种事情发生的几率有多大呢?
我和我妻子携带的基因突变在数千个控制样本中都是独一无二的,因此我们估计这种突变的几率不会大于几千分之一。这意味着随机的基因配对产生基因缺陷宝宝的概率在百万分之一以下。
而宝宝同时携带父母的两个变异基因的概率为四分之一,因此生下基因缺陷宝宝的概率在四百万分之一以下。
如果没有更多的确诊病例,这种基因缺陷的发生概率会继续降低。
我们知道我们小女儿也有四分之一的概率同时携带两个突变的基因。
她一个也没有。
图:Victoria还没有一岁,已经可以推着Bertrand去校车站了:

新的选择
同我的岳父Casanova博士和我们的朋友Ho博士一起,我们开始研究NGLY1酶缺乏的生物学意义。
Ho医生推测,缺乏NGLY1酶可能会导致内质网紧张。
在这种情况下,某些抗氧化剂可能可以缓解这种紧张。
Cadanova博士专注于研究让我能逾越我的基因突变的药,让我失效的基因重新制造这种酶。
在多伦多的研究人员会面后,Casanova博士将注意力放在了庆大霉素上,因为它已经被用于治疗某些形式的囊性纤维化。
合成NGLY酶
就在我们在杜克大学的会面后不久,我妻子自己的研究“中奖了”:杜克大学的小组和犹他大学的小组都还不知道,NGLY1酶的某个变种已经可以合成了。Genzyme公司持有这种合成技术的专利。
它的去糖基的能力在实验室内证实有效,人们在二十年前已经知道如何合成这种酶了。
你可以花244美元去这里订购一批。
下一步
不幸的是,我们不能简单的买这样一批酶给Bertrand注射进去。
我们需要得到FDA的批准,我们将需要Genzyme公司的合作。
尽管他的病是威胁生命的,他的抽搐发作在加重,他继续失去白质,我们需要证明这种治疗方式是安全的。
即使我们通过了这些步骤,我们可能需要调整配方来提高生物利用度。
而且,在那之后还会有未知的未知。
但是,这关系到Bertrand的生命。
所以,这就是我们要去做的。
结语
三年前我的这篇博文中写到:我们当然会为他竭尽全力,如果还不行,我们要尝试不可能的事情。
但是这是什么意思呢?
在我写的关于博士研究的插图说明中,我提到博士研究就是在人类知识的边缘敲出一个小坑。
这篇文章就包含这样的一个小坑,现在科学必要的而且很复杂的小坑。
科学是将未知变为已知的的系统过程。因而从不可能变为可能是必要的。
在写完这些文字的一段时间后,我加上了这样的一幅插画,来强调这个转变的重要性:

边界上有个小坑,我们快到那里了。我们继续要做的,就是继续前进。
相关页面: * 关于博士研究的插图说明