
Normal
Aside from severe jaundice, Bertrand was normal at birth.
For two months, he developed normally.
At three months, his development had slowed, but it was "within normal variations."
By six months, he had little to no motor control.
He seemed, as we described it, "jiggly."
Something was wrong.
"Brain damage"
Bertrand was eight months old when he met with his developmental pediatrician for the first time--just after our move to Utah.
I was at my first faculty retreat on the day of his exam, and after it let out, I found a flood of voicemail and text messages from my wife.
My heart jumped.
The pediatrician thought Bertrand had brain damage, so she scheduled an MRI for the following week.
No brain damage
The MRI showed an apparently healthy, normal brain.
So, his case was escalated to a pediatric neurologist.
The neurologist confirmed that he had a movement disorder, but his presentation was "puzzling": he had neither characteristic chorea nor ataxia.
The neurologist ordered a round of bloodwork.
This was the first of dozens of blood draws to come.
(We now send Bertrand's "favorite" phlebotomists holiday cards.)
The first death sentence
The lab results reported only one anomaly: extremely elevated alpha-fetoprotein (AFP) relative to what it should have been for his age.
Only a handful of known disorders cause elevated alpha-fetoprotein.
Only one of them sits at the intersection of movement disorder and elevated AFP: ataxia telangiectasia (A-T).
A-T is a degenerative, fatal, incurable, untreatable disorder.
My wife and I were heartbroken.
Are you related?
Because A-T is an autosomal recessive genetic disorder, this would be the first of many times that my wife and I were asked:
I'm of Ohio farmland and northern European descent. My wife is multigenerational Puerto Rican.
We can trace our family trees backward for centuries.
No.
We are not related.
Genetics for programmers
[Note: My formal education in biology is two months in high school. Please email me corrections if I made any mistakes.]
To understand why every doctor kept asking us whether we were related and how unlikely Bertrand's final diagnosis is, you need to understand how genes and mutations work.
DNA
Your genome contains the information necessary to build and operate you.
Your genome is transliterated in DNA--a molecular encoding of a language with just four letters: A, C, T and G.
(A, C, T and G stand for adenine, cytosine, thymine and guanine.)
A, C, T and G are to life what 0 and 1 are to computers.
What's important in life, as in computing, is how these sequences encode information or computation.
In computing, a sequence like 00000100 might mean "add in place", so that 00001000 0001 0010 could mean "add the number in register 1 to the number in register 2".
Codons and the standard genetic code
In computing, most computers run on the x86 instruction set.
Remarkably, in life, there is also a dominant instruction set--the standard genetic code, as described in the DNA codon table.
The genetic code is an instruction set for making proteins by chaining together individual amino acids.
The genetic code is made up of instructions called codons.
Each codon is a three-letter sequence in DNA that encodes either an amino acid to insert or the command "stop construction of this protein."
For example, the codon TTG means "insert a Leucine."
With four letters in the alphabet, there are 43 = 64 possible codons, but there are only 25 genetic instructions, since some codons encode the same amino acid and several encode "stop."
Genes
In the human genome, a gene is a functional unit--kind of like the code for a procedure in a program. Each one is composed of exons (codon sequences that are involved in expressing the protein) and introns (ignored sequences that have about the same effect as code comments).
The exons describe a sequence of amino acids to fold into a protein.
When a gene is compiled into a protein--usually as an enzyme--that enzyme acts like a function inside the cell: enzymes enable the reaction of input molecules into output molecules.
With the exception of men and the sex chromosomes, humans have two versions of every gene--one from their mother and the other from their father.
Having two functionally similar versions of each gene provides redundancy.
Mutation and evolution
This redundancy in genes is a powerful protection against mutations.
A mutation is an alteration of an organism's genetic code.
Some mutations change one letter to another, e.g., T to A, or A to G.
This could change the amino acid that is inserted, as in TTT (Phenylalanine) to TTA (Leucine), or not, as in TTA (Leucine) to TTG (Leucine).
Stop mutations
Though uncommon, it is also possible for a mutation to turn a codon into the "stop" instruction, which prematurely terminates production of the protein, e.g., TGG (Tryptophan) -> TGA (Stop).
This is called a nonsense mutation, because the resulting protein is rarely able to perform the task of the original.
Frame mutations
Some mutations insert or delete an arbitrary number of letters. If the number changed is divisible by three, the codons after the insertion or deletion will read correctly; this is called a in-frame mutation.
In-frame mutations may slightly alter the functionality of the protein.
(They can destroy the functionality too.)
If the number changed is not evenly divisible by three, the codons after the insertion or deletion will be garbled. This is a frame-shift mutation.
In most cases, the later in the gene the mutation occurs, the more functional the resulting protein will be. (But, as we eventually learned with Bertrand, even a frame-shift mutation at the very end of a gene sometimes breaks the resulting protein.)
Then what happens?
When a mutation occurs, there are four possibilities for the mutant:
- Nothing happens.
Some mutations don't impact the functionality (or the structure) of the resulting protein. But, even if the mutation breaks the protein, there may be no sign of this. If the redundant version of the gene from the other parent is capable of producing enough of the protein, there are no symptoms. This is what usually happens. Redundancy is great! -
Insufficiency.
If the other gene can't produce enough of the protein, there will be symptoms ranging from the barely perceptible to the severe and/or fatal. -
Active harm.
If the mutant gene produces a new protein which is actively harmful, there will be symptoms. When a single abnormal gene causes problems, it is an autosomal dominant disorder. -
Evolution.
If the mutant gene produces a better, more effective version of the protein, the resulting individual is more "fit." The probability of survival and reproduction increases.
Autosomal recessive disorders
Autosomal recessive disorders like A-T happen when otherwise harmless (but nonfunctional) mutant genes meet back up with themselves.
If two descendants of the original mutant breed, and both carry a copy of the mutant gene, there is a one in four chance that a given child of theirs will inherit both copies of the mutant gene.
This is why many genetic disorders are associated with specific groups or geographies--situations where the likelihood of a common mutant ancestor is increased.
When someone inherits two versions of the same mutant gene, the parents are (usually very distant) cousins.
But, Cristina and I are (demonstrably) not related.
Yet, Bertrand has an autosomal recessive disorder.
Accusations of infidelity
It didn't help that Bertrand looks much more like Cristina than me.
After confident assertions that we were not related, most of Bertrand's new doctors for the next couple years would find a way to pull my wife aside and ask her alone: "Is there any chance he is not Bertrand's father?"
This is not the case.
Into the empty set
Once the shock of the A-T diagnosis wore off, we started researching.
Within days, we were convinced that Bertrand did not have it.
Even though he had elevated AFP and a movement disorder, his presentation differed from the medical literature.
We had the gene test for A-T, and, as we expected, it came back negative.
At 10 months old, the intersection of Bertrand's symptoms landed him in the empty set.
With each new finding, the empty set just kept getting emptier.
Take your best shot
Small findings popped up over the next few months.
Each finding provoked new rounds of hypotheses and (negative) tests.
We found elevated levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST), which pointed to liver dysfunction.
But, a full examination by a gastroenterologist turned up nothing.
Mentally, Bertrand's development halted around 8 months old, and that is where it remains today, even though he is now 4 years old.
The peculiar nature of Betrand's case was attracting attention from specialists that wanted to take a shot a diagnosis.
None succeeded.
The next death sentence
At about 15 months old, the next big finding came.
We found oligosaccharides, chains of simple sugars, in his urine.
This finding immediately implicated a smally family of genetic disorders: inborn errors of cellular metabolism.
Specific groups within this family include oligosaccharidoses, lysosomal storage disorders, congenital disorders of glycosylation and mitochondrial disorders.
(We now know that Betrand created a new category in this family of diseases: a congenital disorder of deglycosylation.)
Bertrand's life expectancy was cut to about two to three years.
We didn't know which one he had, but the (ultra-rare) disorders all have names like:
Alpha-Fucosidosis Alpha-Mannosidosis Alpha-N-Acetylgalactosaminidase Deficiency Aspartylglycosaminuria Beta-Mannosidosis Galactosialidosis Gaucher Disease GM1 gangliosidosis GM2 gangliosidosis GSD II (Glycogen Storage Disease Type II) I-Cell Disease Mucolipidosis II Mucolipidosis III Pompe Disease Pseudo-Hurler Polydystrophy Sandhoff Disease Schindler Disease Sialidosis
Options
With the exception of mitochondrial disorders, these disorders tend to be caused by the inability to produce an enzyme necessary for some component of cellular metabolism.
In theory, introducing this missing enzyme into the cells of the body could stop the progression of the disorder.
In a small number of cases, the enzyme can be synthesized, although delivery of the enzyme to all cells is a complex pharmaceutical challenge.
(It is hard to get many molecules past the blood-brain barrier.)
But, in most cases, humanity doesn't know how to synthesize the enzyme.
As a result, the only chance of delivering the missing enzyme is a bone marrow transplant.
Creating a chimera
In a bone marrow transplant, the stem-cell-producing bone marrow of the recipient is killed off (incidentally, along with most of the rest of the patient) and (largely) replaced with the stem-cell-producing marrow of the donor.
Stem cells are capable of becoming many different kinds of cells, and as such, they play an important role in growth and repair.
As the donor stem cells proliferate, the recipient becomes an artificial chimera: a hybrid organism with cells from two distinct genetic sources.
When the donor cells produce the missing enzyme, it may be enough for the body to function properly, or at least, better.
Duke
Within weeks of finding oligosaccharides, we had begun blood tests to narrow down which specific disorder (and which missing enzyme) it was.
In the mean time, we traveled to Duke University to meet with Dr. Joanne Kurtzberg, the expert in bone marrow stem cell transplants for the treatment of inborn errors in metabolism.
Before risking Bertrand in a stem cell transplant (a roughly 30% mortality rate), Dr. Kurtzberg wanted to know exactly which disorder he had.
So, we met with the geneticists Dr. Vandana Shashi and Kelly Schoch.
We've been working with these two and their team ever since.
Epilepsy and white matter loss
At Duke, the neurology team conducted an EEG and another MRI.
They found "strange," "probably epileptic" activity raging in his brain.
(To this day, his EEGs provoke a crowd of onlookers.)
The MRI showed that his brain had delayed myelination.
His brain was losing (or not gaining) white matter (or, functionally speaking, the networking infrastructure).
This finding was consistent with leukodystrophy--often brought on by inborn errors of cellular metabolism.
Out of options
Before we left Duke, Dr. Kurtzberg told us, frankly yet compassionately, that whichever disorder Bertrand had, he had progressed too far to benefit from a bone marrow transplant.
We were crushed.
Focusing on treatment
After Duke, we shifted some energy from diagnosis to treatment.
Discovering that many of Bertrand's abnormal movements were actually seizures was disconcerting.
In fact, Bertrand had three kinds of seizures: myoclonic "jerking" seizures, absence "blank stare" seizures and atonic "drop" seizures.
Within a few months, he began experiencing tonic seizures with prolonged, painful whole-body muscle contractions.
Bertrand started Keppra, a widely prescribed antiseizure medication.
Ketogenic diet
When Keppra proved only partially effective at controlling his epilepsy, we tried the ketogenic diet.
The ketogenic diet is a strict high-fat diet which forces the brain to switch from glucose to ketone bodies for its primary fuel.
On the ketogenic diet, for every one gram of carbohydrates and/or protein Bertrand ate, Bertrand ate an additional four grams of fat.
The diet has been widely studied and prescribed, but much is unknown as to why it works in many cases of intractable epilepsy.
On the diet, Bertrand's drop seizures stopped almost entirely, and the remaining types of seizures diminished greatly.
But, when the tonic seizures set in, we started looking for more options.
No tears
Sometimes a symptom is too obvious to notice.
But, at nearly two years old, we noticed Bertrand never had tears.
He could cry.
But he never made tears.
A quick google search for alacrima found Allgrove syndrome.
By now, we'd tested for and exhausted all known inborn errors of cellular metabolism, so we were eager to follow the lead.
To the NIH
Cristina contacted Dr. Stratakis at the NIH, a specialist in Allgrove.
Bertrand's unusual presentation piqued Dr. Stratakis's interest, so he had Cristina and Bertrand fly out to see a panel at the NIH.
The panel guessed that Bertrand likely did not have Allgrove, but that it would be worth a genetic test.
The genetic test was negative.
The panel at the NIH finally concluded that Bertrand might have male Rett syndrome or possibly Schinzel–Giedion.
While phenotypically similar to male Rett, further testing showed that he had neither disorder.
The nuclear option: ACTH
As Bertrand's wrenching tonic seizures increased in intensity and frequency, we became desperate to stop them.
In some cases of intractible epilepsy, high doses of ACTH halt them.
In a lucky break, ACTH worked for Bertrand.
But, the twice-daily hormone injections had side effects.
He bloated to double his weight.
His hair thinned and balded.
He grew facial hair.
He was also in a permanent state of rage.
Imagine taking a rocket-sled through puberty at two years old.
But, the seizures were gone.
Near death experience
The end of both ACTH and the ketogenic diet came suddenly.
ACTH stripped away Bertrand's immune system.
After a couple months, he contracted a severe respiratory infection.
His tiny two-year-old frame was so filled with fluid that he could not move.
Each segment of his body swelled; he looked like a balloon animal.
We thought he was going to die.
To save him, he was pulled off both ACTH and the ketogenic diet and plugged into a series of tubes, wires and antibiotics.
He looked like the borg.
Laughter
The morning after Bertrand was weaned from ACTH and the ketogenic diet, we heard something we hadn't heard before: laughter.
Still bloated and near death, in his hospital bed, he was laughing.
Everytime the laugh track came on the hospital TV, he chipped in.
It was the most direct sign of Bertrand's humanity we had ever seen.
Cristina was in tears.
The eye of the storm
After Bertrand recovered, his seizures stayed away for almost two months.
Even his EEG looked "normal."
Free of seizures, Bertrand began to learn and develop.
For Cristina and me, they were the best two months of our lives.
Two months post-ACTH, Bertrand laughed at movies.
Seizures return
Eventually the myoclonic seizures returned, but the atonic seizures, absence seizures and tonic seizures did not.
Adding lamictal to his medications dampened the myoclonic seizures, but it made him groggy.
Our quest to find a diagnosis continued.
Liver fibrosis
Since Bertrand's liver values had remained consistently elevated, we agreed to a liver biopsy while he was in the hospital.
The biopsy ruled out two recent considerations: Lafora disease and Unverricht Lundborg disease.
Unfortunately, the biopsy found fibrosis in Bertrand's liver.
His gastroenterologist predicted that his liver would eventually develop cirrhosis and fail.
He recommended ursodiol, so we gave it a shot.
Heart problems: Long QT syndrome
During Bertrand's hospital stay, an electrocardiogram revealed long QT syndrome.
Long QT is a rare heart condition which can lead to fatal irregularities in heart rhythm.
Long QT usually has a genetic basis, but it can also be drug-induced.
For a brief period, we began investigating congenital channelopathies as a possible cause of his troubles.
During this time, there seemed to be a grim race between Bertrand's brain, heart and liver to kill him.
A dangerous hypothesis
With almost every avenue for diagnosis exhausted, Cristina and I had formulated hypotheses about Bertrand's condition.
Given the unrelatedness of our families (and hence the unlikeliness of an autosomal recessive disorder) and the lack of any history of genetic diseases on either side, we felt that Bertrand's condition was likely the result of a de novo mutation.
We started to assume that Bertrand, not we, had a unique mutation.
As such, we assumed there would be no risk to having another child.
We were wrong.
But, assuming Bertrand had a novel mutation, we hatched a plan to find it.
Dinner with a geneticist
I managed to get a dinner with University of Utah geneticist Dr. Lynn Jorde.
I asked him about the possibility of sequencing three genomes: mine, my wife's and Bertrands.
Genomic sequencing yields the genetic code of an organism--about 3.1 billion letters in the case of humans.
Presumably, if we had these three sequences, we could find the few mutations where Bertrand differed from both of us.
But, this is easier said than done, and once done, it's still not that easy.
Error rates in sequencing
The sequencing process has an error rate.
That is, any given sequencing would contain a few false mutations.
With an error rate of 1 in 10,000, a given sequencing will have about 310,000 errors--false mutations.
Repeated sequencing drops the error rate, but drives up the cost.
At the dinner table, I estimated it would cost about $500,000 to achieve a "reasonable" level of confidence.
Dr. Jorde nodded apologetically in agreement.
And then what?
But, suppose we found the mutations; then what?
We'd have to investigate each one to see how it impacted protein construction and function--a daunting task.
But, theoretically, it could be done.
An opportunity: Exome sequencing
Throughout Bertrand's ordeal, Dr. Shashi and Kelly Schoch at Duke had stayed in touch and worked with us to test hypotheses.
They felt strongly that Bertrand had an undiscovered genetic disorder.
And they devised a clever way to test that hypothesis.
They proposed using a new technique--whole-exome (instead of whole-genome) sequencing--on the three of us.
Only about 2% of our DNA--the exome--actively codes proteins.
It's estimated that mutations in this 2% are responsible for the vast majority of genetic disorders.
Whole-exome sequencing can economically sequence this small fraction of our DNA.
If the mutation was in Bertand's exome, we would be able to find it.
Bertrand, and 11 other undiagnosed children, joined a pilot study at Duke.
Two bullets dodged
After we began treating Bertand's liver with ursodiol and closely monitoring signs of liver distress, we saw steady improvement.
A year later, all of his liver values entered the "normal" range.
Bertrand would not die of liver failure.
Repeated testing of his heart allowed his cardiologist to conclude that that Bertrand had drug-induced (not congential) long QT syndrome.
In Bertrand's case, the cause of long QT turned out to be erythromycin, which had been used to treat his infection in the hospital.
Corneal erosion
Right after he was cleared of long QT and liver failure, Bertrand started having serious eye infections.
One failed to respond to antibiotics, and it required surgery to drain the pus from his cornea.
The lack of tears and low moisture meant that his cornea was eroding.
Scars formed just below his pupils--clouding but not obscuring his vision.
With the help of his opthalmologist, we started a regimen that required putting lubricating ointment and drops in his eyes every two hours.
Since we launched this regimen, his eyes have improved, but forgetting his drops for even half a day usually results in week-long eye infection and more scarring.
Bertrand cannot be without a trained, trusted caregiver for more than a couple hours.
Pregnancy
About a month after the exome experiment at Duke launched, Cristina was pregnant with our second child.
We realized that there was a chance that the experiment would reveal that our unborn child had (or could have) the same disorder as Bertrand.
We decided that, regardless of what we learned, we would not use that information to direct the course of the pregnancy.
(The IRB protocol dictated that pregnant families were to be excluded from the experiment because of exactly this thorny ethical issue.)
Stem cells
While the exome experiment ran, we continued to treat Bertrand's epilepsy.
One hypothesis at the time was that Bertrand was not a full mutant--that when he was no more than a handful of cells, one of those cells mutated.
The term for this condition is somatic mosaicism, since the individual is a hybrid of two very similar genetic sources.
Operating under the assumption of this possibility, we decided to gamble.
We had banked Bertrand's cord blood stem cells at birth.
Dr. Kurtzberg was running an experimental study at Duke testing the impact of stem cell infusion (not transplantation) on children with conditions that impacted the brain (like cerebral palsy).
Since infusions of one's own stem cells would be harmless, we gave it a shot.
Our thought was that if the mutation did not affect his cord blood, these stem cells might begin to help repair some of the brain damage and produce a functional version of his missing enzyme.
Even if it didn't produce the enzyme, there was reason to hope that it might temporarily halt or reverse the loss of white matter in his brain.
To Cleveland Clinic
Given the multifocal nature of Bertrand's epilepsy, we had been told that surgery was not an option.
But, to be absolutely sure, we headed to Cleveland Clinic, the leader in surgical treatment of pediatric epilepsy.
Even though Cleveland Clinic confirmed that Bertrand was not a candidate for surgery, they did augment his medications to reduce his seizures substantially.
But the MRI at Cleveland showed something stunning: the loss of white matter in Bertrand's brain had (temporarily) plateaued.
By itself, this is not enough to claim that the stem cell infusion had worked, but it is encouraging, and it suggests that further research into stem cell therapy is warranted.
The first mutation
At three and a half years old, a week before Bertrand's sister Victoria was born, we got a call from Dr. Shashi's team at Duke.
Due to the IRB protocol, they could not reveal what they had found, but they felt strongly that Victoria could not be impacted by Bertrand's disorder.
They wanted blood samples from Cristina's parents (but not mine).
We put it together instantly: the Duke team found a mutation in the X chromosome that they thought was responsible for Bertrand.
When the X chromosome contains a mutation, it is often the case that women are not fully affected: they have a redundant X chromosome to compensate.
When men have a mutant X chromosome, the lack of redundancy can cause severe symptoms.
The takeaway is that if Bertrand had an X-linked condition, our daughter Victoria might be a carrier, but should be no less functional than Cristina.
We were overjoyed.
But, the team at Duke had it wrong.
The wrong mutation
The test results for Cristina's parents came back after Victoria was born.
Cristina's father had the same mutation in his X chromosome.
Even though this X-linked mutation was unique to the control database of over a thousand genomes and it impacted a protein that could have plausibly explained some of Bertrand's symptoms, the fact that Cristina's father was "normal" ruled out its culpability.
Vitamin deficiency
Sometimes, a solution seems almost too obvious to try.
In Bertrand's case, vitamins were one of those solutions.
While reading up on causes for dry eyes, Cristina stumbled across vitamin deficiencies.
Even though Bertrand was taking a daily multivitamin, testing Bertrand's vitamin levels revealed serious deficiencies in the vitamins stored in the liver.
With large daily megadoses of these vitamins, we brought his levels into the "normal" range.
Since then, Bertrand has been able to cry tears--not much--but enough.
Unfortunately, because of the corneal scarring, he no longer blinks, so he still requires frequent ointment and lubrication--but much less than before.
Diagnosis: N-Glycanase 1 deficiency
At four and a half years old, we got the call from Duke.
The exome study had concluded, and they had the answer.
Cristina and I each carried a different mutant NGLY1 gene.
I have a nonsense (stop) mutation in exon 8; Cristina has a frame-shift in the last exon.
Cristina and I each produce about half the amount of N-Glycanase 1 as the regular population. Bertrand got both mutant NGLY1 genes, leaving him with no ability to produce the enzyme:
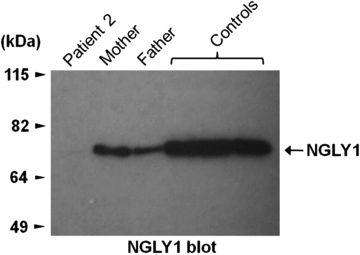
The dark bands in this blot indicate the amount of N-Glycanase 1.
Bertrand is "Patient 2."
He is the first and so far only human being known to lack this enzyme:
N-Glycanase 1 plays an important role in deglycosylating misfolded proteins, allowing them to be recycled into their constituent amino acids.
Bertrand's cells appear to be accumulating misfolded glycoproteins.
The odds
It's worth pondering the odds of this diagnosis.
Since the mutations carried by Cristina and me were both unique among a control population of thousands, we can estimate that no more than one in a thousand has these mutations.
That means that a random coupling has at best a one in a million chance of being capable of producing a Bertrand.
But, since only one in four children from that coupling would have both genes, and hence, the disorder, at most one in four million births should produce a Bertrand.
Until more people with similar NGLY1 mutations are found, the upper bound on the probability of creating a Bertrand will continue to shrink.
We now know that there was a one in four chance that Victoria would inherit both mutations.
She has neither.

Not even a year old, Victoria pushes Bertrand to the schoolbus stop.
New options
Working with Cristina's father Dr. Manuel Casanova and our friend Dr. Karen Ho, we started to sort through the biological implications of N-Glycanase 1 deficiency.
Dr. Ho hypothesized that the lack of N-Glycanase 1 could cause stress on the endoplasmic reticulum.
In this case, certain antioxidants may be helpful in reducing that stress.
Dr. Casanova focused on pharmaceuticals which might be able to "hack" my stop mutation, causing an occasional read-through of the mutation that would allow my broken copy of NGLY1 to produce N-Glycanase 1.
After meeting with a researcher in Toronto, Dr. Casanova settled on gentamicin as a promising candidate, since it is already being used in that capacity to treat some forms of cystic fibrosis.
Synthesizing N-Glycanase
But, a couple days after our meeting at Duke, Cristina's own research hit the jackpot: unbeknownst to the team at Duke and our team in Salt Lake, one variant of N-Glycanase 1 is already synthesizable.
(Genzyme holds the patent on its synthesis.)
Its deglycosylating ability is useful in laboratory settings, and humans have been able to make the stuff for about two decades.
You can order a batch for $244.
Next steps
Unfortunately, we can't just order a batch and inject Bertrand.
We need to get FDA approval, and we'll need Genzyme's cooperation.
Even though his disease is life-threatening, his seizures are worsening and he continues to lose white matter, we'll need to prove that it's safe.
Even once we're through those steps, we may need to tinker with the formulation to improve bio-availability.
And, there are certainly unknown unknowns beyond that.
But, Bertrand's life is on the line.
So, that's what we'll do.
Epilogue
On the somber blog post announcing the discovery of oligosaccharides three years ago, we concluded with a promise to Bertrand:
And, if that fails, we'll try the impossible.
But, what does it mean to do the impossible?
In The Illustrated Guide to a Ph.D., I talked about making dents in the boundary of human knowledge.
This article contains the story of one such dent--of the messy but essential process of modern science.
Science is the systematic transformation of the unknown into the known.
It is necessarily then a transformation of the impossible into the possible.
Some time after the guide was written, I added an epilogue emphasizing the importance of that transformation:

There is a new dent in the boundary.
We're almost there.
All we need to do is keep pushing.
Update: Two years after this was written, Seth Mnookin penned the penned the sequel for The New Yorker.